


锂电池研究中理论模拟的三个方面
宏观尺度:有限元方法等
介观尺度:相场模拟、分子力场等
原子尺度:第一性原理、密度泛函理论、分子动力学、蒙特卡罗等
根据所研究问题的内容与所在的空间与时间尺度,计算材料学的模拟方法涵盖了从微观原子、分子水平,到介观微米级别,直至宏观尺度的各种理论。下图展示了计算材料学在不同空间与时间尺度上的模拟方法
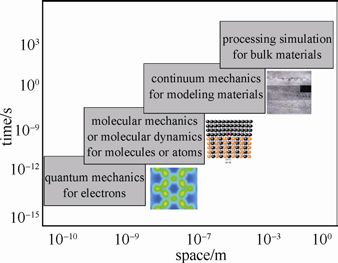
基于密度泛函理论的第一性原理计算同分子动力学、蒙特卡罗方法相结合,在材料设计、合成、模拟计算和评价诸多方面有许多突破性的进展,已经成为原子尺度上材料计算模拟的重要基础和核心技术。
第一性原理计算的宗旨是不采用任何经验参数,只采用电子质量、光速、质子质量、元电荷、普朗克常数5 个基本物理量计算研究微观粒子的行为。
密度泛函理论的发展
20 世纪60年代,密度泛函理论建立。
1985年,Car和Parrinello 两人把分子动力学方法和第一性原理计算结合起来,使得第一性原理方法在处理问题时变得更为实际。
1998年,Kohn 教授因其对密度泛函理论(DFT)发展所做出的巨大贡献荣获了该年度的诺贝尔化学奖。
随后,计算材料学特别是基于密度泛函理论的第一性原理计算在材料科学研究中迅速升温,已成为当今计算材料科学的一个最为重要的工具。
基于密度泛函理论的第一性原理
第一性原理计算方法有着半经验方法不可比拟的优势,只需知晓构成微观体系的元素及其位置,即可以应用量子力学来计算出该微观体系的总能量、电子结构等物理性质。
1)现象解释:第一性原理计算可作为真实实验的补充,通过计算可以更为深入地理解被模拟体系的特征和性质;
2)理论预测:通过第一性原理计算可以在尚无实验的情况下对研究体系进行预测和设计。
锂电池研究中,最常见的模拟类型
电池电压、嵌锂电位、电极材料的电子结构、能带结构、弛豫结构、迁移路径、缺陷生成能、活化能、锂离子传输动力学和脱嵌锂相变、材料中温度场分布、应力场分布等。
锂电池中尚不明朗的基础科学问题有哪些?
如SEI的生长机制、离子在电极材料中的扩散动力学特性、电极材料充放电过程中结构的演变、电位与结构的关系、空间电荷层分布等。
为什么理论计算的嵌锂电位比实际低?
问题:研究早期人们发现,几乎对所有的正极材料,密度泛函理论计算得到的电压都偏低。
解释:麻省理工学院G. Ceder研究组认为这是由于含有3d过渡金属离子的正极材料属于强关联电子体系,其3d轨道的电子是高度局域化的。而在单电子近似的密度泛函理论框架下,局域化的电子会引入自相互作用,因此导致嵌锂电位被低估。
解决:通过对标准的GGA 进行+U修正,也即 GGA+U的方法,或采用杂化泛函HSE06,计算得到了与实验值更为接近的电压,见下图。
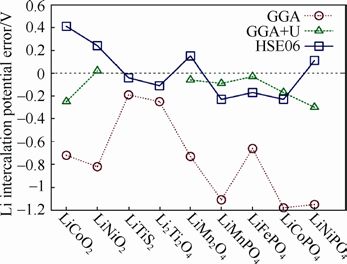
标准GGA与GGA+U、HSE06计算得到各种锂离子电池电极材料嵌锂电压
在含有3d 过渡金属离子的氧化物正极材料计算中,强关联效应已经公认为需要考虑的效应。
相关学习资料:
[1] Zhou F,Cococcioni M,Marianetti C A,et al. First-principlesprediction of redox potentials in transition-metal compounds with LDA + U[J].Phys. Rev. B,2004,70(23):235121.
[2] Chevrier V L,Ong S P,Armiento R,et al. Hybrid density functionalcalculations of redox potentials and formation energies of transition metalcompounds[J]. Phys. Rev. B,2010,82(7):75122.
计算电极材料的稳定性
案例一:计算分析脱嵌锂过程中正极材料的分解问题
麻省理工学院G. Ceder研究组计算了LixCoO2、LixNiO2、LixMn2O4材料在不同Li含量下的热力学相图,阐明了这3 种材料的分解机制。
以 LiNiO2为例,计算了该材料在0 K、220 K、730 K 的Li-Ni-O2三元相图(见下图)。
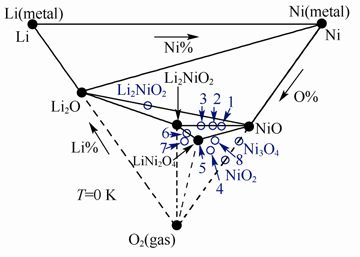
计算得到的Li-Ni-O2体系在0K时的三元相图
通过分析相图中LixNiO2 稳定存在的区域及分解所经过的区域,认为LixNiO2的分解是通过层状向尖晶石相的放热反应和尖晶石向岩盐相的吸热反应两步来实现的。
相关学习资料:
[1] Wang L,Maxisch T,Ceder G. A first-principles approach to studying the thermalstability of oxide cathode materials[J]. Chem. Mater.,2007,19(3):543-552
案例二:模拟分析循环过程中氧析出问题
电极材料在循环过程中的不稳定性还可能以氧析出的形式表现出来,这种现象在富锂相正极材料中较为常见。
富锂锰基xLi2MnO3·(1−x)LiMO2(M=Ni、Co、Mn等)正极材料循环过程中释放氧气、电子电导低等问题导致其不可逆容量高、倍率性能和循环性差,阻碍了该类材料在实际中的应用。
中科院物理所陈立泉院士团队对此进行了深入研究,通过第一性原理计算系统地研究了富锂锰基正极材料的母相化合物Li2MnO3晶格中氧在不同锂含量时的稳定性。下图为通过第一性原理计算得到的不同脱锂态Li2−xMnO3氧气释放过程中的反应焓和吉布斯自由能。
可知,当脱锂量x≥0.5 时会有氧气产生,进而导致结构的变化。
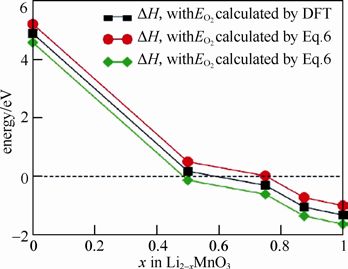
计算得到的不同脱锂态Li2−xMnO3氧气释放过程中反应焓和吉布斯自由能
相关学习资料:
[1] Xiao R J,Li H,Chen L Q. Density functional investigationon Li2MnO3[J]. Chemistry of Materials,2012,24(21):4242-4251
案例三:计算模拟掺杂对稳定性的影响
中科院物理所陈立泉院士团队通过第一性原理计算研究了不同晶格位Mo 掺杂Li2MnO3晶体的结构稳定性及Li+在晶格中不同跃迁方向的迁移能垒。下图是采用PBE+U交换关联势计算得到的不同脱锂态的LiyMnO3和LiyMn0.75Mo0.25O3的氧气释放过程中反应焓和吉布斯自由能。
可知,与未掺杂的Li2MnO3相比,掺Mo的Li2MnO3结构稳定性有了大幅度提升。

计算得到的不同脱锂态LiyMnO3和LiyMn0.75Mo0.25O3的气释放过程中反应焓和吉布斯自由能
相关学习资料:
[1] Gao Y R,Ma J,Wang X F,et al.Improved electron/Li-ion transport and oxygen stability of Mo-doped Li2MnO3[J].Journal of Materials Chemistry A,2014,2(13):4811-4818.
计算电子结构与电化学性能的关系
电池材料的电子结构也与材料的电化学性能有着密切的关系:
1)电极材料中,脱嵌锂过程中电荷补偿的来源和氧的稳定性与过渡金属及氧的分波态密度的相对位置有关;
2)电极与电解质界面的化学稳定性则可从能态密度中做出定性预测,此外电池的倍率性能也与电极的电子导电性有关;
3)电池中的固态电解质材料需要具有电子绝缘的特性,材料的能隙宽度与其电化学窗口的宽度有关。
电极与电解质之间的界面问题一直是电池研究中的难点。
美国维克森林大学A. W. Holzwarth团队尝试通过理论计算理解Li3PS4和Li3PO4与金属Li负极之间的化学稳定性。以下两图分别为计算得到的β-Li3PO4、γ-Li3PS4与金属Li界面模型的分波态密度图。
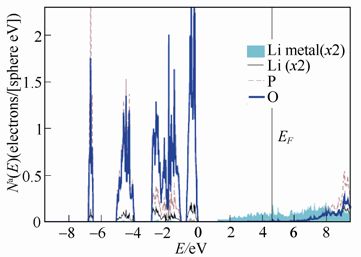
β-Li3PO4与金属Li界面模型的分波态密度图
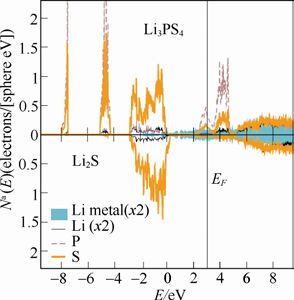
γ-Li3PS4与金属Li界面模型的分波态密度图
图中显示,当β-Li3PO4与金属Li接触时,金属Li的电子态分布在P和O的占据态的上方,与P、O原子之间难以相互作用,因此β-Li3PO4/Li界面的化学稳定性较高;而γ-Li3PS4/Li 的界面上,Li 的电子态与S的电子态之间有显著地重叠,二者之间容易发生电子转移,形成Li-S键,造成界面层形成新相,从而引起更为复杂的电化学过程。
相关学习资料:
[1] Goodenough J B,Kim Y. Challenges forrechargeable Li batteries[J]. Chemistry of Materials,2010,22:587-603.
[2] Lepley N D,Holzwarth N A W,Du Y A. Structure, Li+ mobilities, and interfacial properties ofsolid electrolytes Li3PS4 and Li3PO4 from first principles[J]. Phys. Rev. B,2013,88:104103.
[3] Shi S Q,Liu L J,Ouyang C Y,et al. Enhancement of electronicconductivity of LiFePO4 by Cr doping and its identification by first-principlescalculations[J]. Phys. Rev. B,2003,68(19):195108.
[4] Quartarone E,Mustarelli P.Electrolytes for solid-state lithium rechargeable batteries:Recent advances and perspectives[J]. Chem. Soc. Rev.,2011,40(5):2525-2540.
模拟离子输运机制
锂离子的传输是锂二次电池中核心的输运过程,锂离子传输的路径、能量势垒等与电池的倍率性能、极化程度、离子互占位等现象有着直接的联系。
锂离子在材料中的扩散性质一方面可以通过基于过渡态理论的弹性能带方法(NEB)获得,另外也可以 采用基于第一性原理的分子动力学方法计算得到。
案例一:证明LiFePO4中Li+是一维扩散
中国科学院物理研究所固态离子学实验室 C Y Ouyang等通过第一性原理计算研究了锂离子在橄榄石结构的LiFePO4中的扩散机理。
通过对不同的可能扩散路径的迁移势垒的计算,表明对于LiFePO4、FePO4 及Li0.5FePO4 来说,扩散势垒沿着c轴方向分别为0.6、1.2 和1.5 eV。而在沿着a 轴、 b 轴方向的迁移路径上,其扩散势垒非常大,以至于锂离子难以扩散。这说明锂离子在LiFePO4 中的扩散是一维的。
案例二:探究LiFePO4中的Li位掺杂
为了进一步解释Cr在Li 位掺杂的LiFePO4材料电子电导率得到了大幅度提高而其电化学性能却没有得到明显改善的现象
他们计算研究了纯的和Li位掺Cr的LiFePO4中锂离子和铬离子沿着一维扩散通道输运的能量势垒(下图所示分别为Li与Cr沿着C轴方向的迁移势垒)
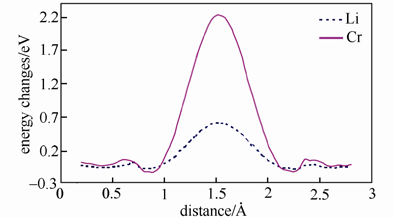
Li与Cr在LiFePO4晶体中沿着c 轴方向的迁移势垒
结果发现锂离子可以很容易地沿着扩散通道扩散,但是铬离子很难离开本来的位置。这意味着铬离子堵塞了材料的一维扩散通道,见下图。
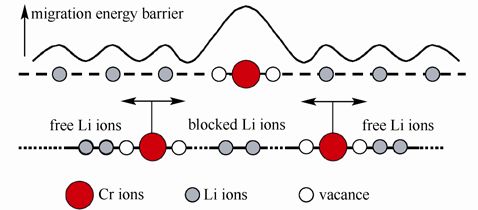
Cr 掺杂导致的锂离子运输阻塞效应模型
从不阻塞一维离子输运通道的角度出发,他们考虑了低价态Na+的Li位掺杂以及其它金属离子的Fe位掺杂对材料动力学性能的影响,并发现两种方法都能够不同程度地提高材料的电子电导率,且不会阻塞Li+的一维输运通道。
相关学习资料:
[1] Ouyang C Y,Shi S Q,Wang Z X,et al. The effect of Cr doping onLi ion diffusion in LiFePO4 from first principles investigations andMonte Carlo simulations[J]. Journal of Physics:CondensedMatter., 2004,16(13):2265-2272.
[2] Ouyang Chuying(欧阳楚英). 锂离子电池正极材料离子动力学性能研究[D]. Beijing:Institute of Physics ChineseAcademy of Sciences,2005.
[3] Ouyang C Y,Shi S Q,Wang Z X,et al. First-principles study of Liion diffusion in LiFePO4[J]. Phys. Rev. B,2004,69(10):104303
模拟缺陷生成能对电导率的影响
材料中的缺陷和杂质会改变材料的许多性质,包括电子电导、离子电导等与锂电池性能紧密相关的性质。
案例一:模拟缺陷对电导率的影响
美国海军研究实验室Michelle D.Johannes团队通过第一性原理计算研究了橄榄石结构的LiFePO4中的本征缺陷及掺杂对其电子传导和离子传导的影响。
计算结果表明,在LiFePO4本征的锂空位缺陷和空穴小极化子分别决定着材料的离子输运和电子输运过程。对一价掺杂元素(Na、K、Cu、Ag),二价掺杂元素(Mg、Zn),三价掺杂元素(Al),四价掺杂元素(Zr、C、Si)和五价掺杂元素(V、Nb),他们分别计算了各种元素掺入不同晶格位置的缺陷形成能,不同合成条件对掺杂浓度的调节以及掺杂后对体系输运性质的改变,发现通过缺陷控制的合成手段可能有效地改善体系的电子导电性。
相关学习资料:
[1] Hoang K,Johannes M D. First-principles studies of the effects of impuritieson the ionic and electronic conduction in LiFePO4[J]. J. PowerSources,2012,206:274-281.
晶体结构及演化
电极材料在脱嵌锂的过程中,有时会出现一些有趣的结构演化过程,如在石墨嵌锂过程中出现的“阶”结构。
近年来,随着透射电镜技术的发展,人们在LiFePO4的脱锂样品中观察到了三相界面, 并且也发现了“阶”的现象。
案例一:计算模拟正极材料脱锂过程的结构变化
中科院物理所黄学杰等通过第一性原理计算研究了LiFePO4脱锂过程中出现的“阶”结构,见下图。
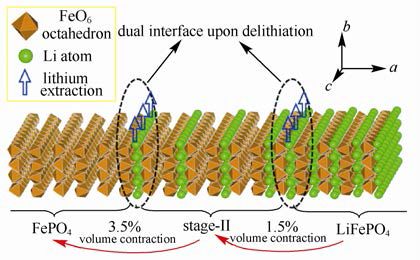
LiFePO4充放电过程中形成的“阶”结构模型
结果显示,“阶”结构是一种受Li+传输动力学控制的热力学亚稳态。其成因在于,当一层锂脱出后其最近邻锂层的Li+跃迁受到阻碍,因此充电过程中倾向于隔层脱锂而不是顺序脱锂。。
相关学习资料:
[1] Sun Y,Lu X,Xiao R J,et al. Kinetically controlledlithium-staging in delithiated LiFePO4 driven by the Fe center mediatedinterlayer Li-Li interactions[J]. Chem. Mater.,2012,24(24):4693-4703.
Si 负极膨胀的力学性能模拟
基于密度泛函理论的第一性原理能够通过求解力矩阵,获得材料的弹性常数,从而得到体模量、剪切模量、杨氏模量、泊松比等一系列力学性能的数值。
大多数电极材料在反复的脱嵌锂过程中会经历体积的变化,如层状材料在脱锂后会出现沿C方向的膨胀,Si 负极在嵌锂之后则会出现多达300%的体积膨胀。
案例一:模拟Si负极的嵌锂膨胀力学性能
美国布朗大学V.B.Shenoy团队计算了不同Li含量的Li-Si合金的学性质,建立了Si 负极嵌锂过程中形变与断裂过程的力学模型。通过计算晶态与非晶态Li-Si合金相的杨氏模量、剪切模量、体模量和泊松比,见下图。发现随着锂浓度的增加,上述模量值几乎呈线性降低。
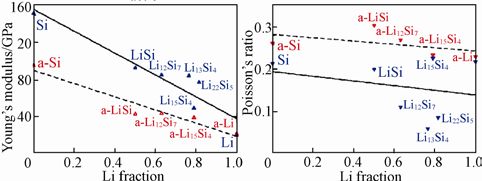
计算得到的晶态(蓝色点)与非晶态(红色点)Li-Si 合金LixSi 在不同Li 含量时的
(a)体模量;(b)剪切模量;(c)杨氏模量;(d)泊松比
表明嵌锂引起了材料的弹性软化,同时也说明了材料的力学特性在电池的电化学过程中不是保持不变的,应力场的分布与形变及裂纹的扩展都与充放电状态相关。
相关学习资料:
[1] 黄杰,凌仕刚, 王雪龙, 等. 锂离子电池基础科学问题 (XⅣ)——计算方法[J]. 储能科学与技术, 2015 (2): 215-230.
[2] Shenoy V B,Johari P,Qi Y. Elastic softening of amorphous and crystalline Li-Si Phaseswith increasing Li concentration : A first-principlesstudy[J]. J. Power Sources,2010,195(19): 6825-6830.
[3] Frenkel D , Smit B. UnderstandingMolecular Simulation[M]. Newyork:Academic Press,2001.
更多学习资料:
[1] Jian Zengyun(坚增运),Liu Cuixia(刘翠霞),Lv Zhigang(吕志 刚). Computational MaterialsScience(计算材料学)[M]. Beijing:ChemicalIndustry Press,2012.
[2] Hafner J,Wolverton C,Ceder G. Toward computational materials design : The impact of density functional theory on materials research[J].Mater. Res. Soc. Bull.,2006,31(9):659-667.
[3] Wang C Y,Zhang X. Multiscalemodeling and related hybrid approaches[J]. Current Opinion in Solid State &Materials Science, 2006,10(1):2-14.
[4] Meng Y S,Dompablo M E. Fristprinciples computational materials design for energy storage materials inlithium ion batteries[J]. Energ. Environ. Sci.,2009,2(6):589-609.
[5] Car R,Parrinello M. Unifiedapproach for molecular dynamics and density-functional theory[J]. Phys. Rev.Lett.,1985,55(22): 2471-2474.
[6] Kohn W. Nobel lecture : Electronic structure of matter—Wave functions and densityfunctionals[J]. Rev. Mod. Phys.,1999,71(5): 1253-1266.
[7] Shi Siqi(施思齐). 锂离子电池正极材料的第一性原理研究[D]. Beijing:Institute of Physics ChineseAcademy of Sciences,2004.
[8] Born M,Huang K. DynamicalTheory of Crystal Lattices[M]. Oxford:OxfordUniversities Press,1954.
[9] Thomas L H. The calculation of atomic fields[J].Proc. Cambridge Phil. Soc.,1927,23(5):542-548.
[10] Fermi E. Un metodo statistico per laDeterminazione di alcune prioprietà dell’Atomo[J]. Rend. Accad. Naz. Lincei,1927,6:602-607.
[11] Hohenberg P,Kohn W. Inhomogeneouselectron gas[J]. Phys. Rev., 1964,136(3B):864-871.
[12] Kohn W,Sham L J.Self-consistent equations including exchange and correlation effects[J]. Phys.Rev.,1965,140(4A):1133-1138.
[13] Slater J C. A simplification of theHartree-Fock method[J]. Phys. Rev.,1951,81(3):385-390.
[14] Ceperley D M,Alder B J. Ground-stateof the electron-gas by a stochastic method[J]. Phys. Rev. Lett.,1980,45(7):566-569.
[15] Perdew J P,Zunger A.Self-interaction corrction to density-functional appoximations formany-electron systems[J]. Phys. Rev. B,1981,23 (10):5048-5079.
[16] Jones R O,Gunnarsson O. Thedensity functional formalism, its application and prospects[J]. Rev. Mod. Phys.,1989,61(3):689-746.
[17] Langreth D C, Perdew J P. Theory ofnonuniform electronic systems.1.analysis of the gradient approximation and ageneralization that works[J]. Phys. Rev. B,1980,21(12):5469-5493.
[18] Becke A D. Density-functionalexchange-energy approximation with correct asymptotic-behavior[J]. Phys. Rev. A,1988,38(6): 3098-3100.
[19] Perdew J P,Chevary J A,Vosko S H,et al. Atoms,molecules,solids,and surfaces—Applications of the generalized gradient approximation forexchange and correlation[J]. Phys. Rev. B,1992,46(11): 6671-6687.
[20] Stadler R,Wolf W,Podloucky R,et al. Ab initio calculations ofthe cohesive, elastic, and dynamical properties of CoSi2 bypseudopotential and all-electron techniques[J]. Phys. Rev. B,1996, 54(3):1729-1734.
供稿 | 深圳市清新电源研究院
部门 | 媒体信息中心科技情报部
撰稿人 | 材料小兵
主编 | 张哲旭

清新电源投稿通道(Scan)
欢迎读者加入
清新电源官方QQ:86931315
▶ 添加备注:姓名+单位+研究方向
▶ 清新电源-文献互助群 608599704
▶ 清新电源-学术交流群 332591118

本文由清新电源原创,作者材料小兵,转载请申请并注明出处:http://www.sztspi.com/archives/9946.html